Pulmonary Arterial Hypertension (PAH) is a rare blood vessel disorder of the lung in which the pressure in the pulmonary artery (the blood vessel that leads from the heart to the lungs) rises above normal levels and may become life threatening.
In the advanced stage of the disease, the blood vessels in the lungs are narrowed, thickened and stiff, causing difficulty for the right heart ventricle heart to pump blood forward, eventually leading to causing the right-sided heart muscle to weakening and progressive right heart failure.
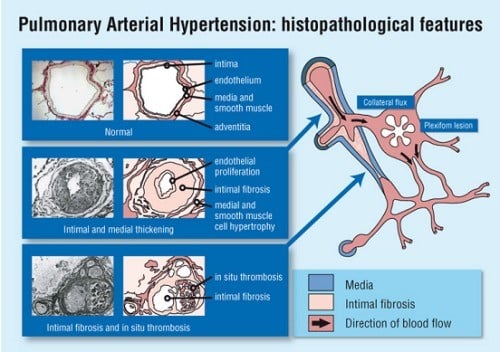
When pulmonary hypertension occurs in the absence of a known cause, it is referred to as idiopathic pulmonary arterial hypertension (IPAH). IPAH is extremely rare, occurring in about 2 to 10 persons per a million population per year. Causes of pulmonary arterial hypertension include collagen vascular diseases (e.g. scleroderma, CREST syndrome or systemic lupus erythematosus), congenital heart diseases (shunts like ventricular and atrial septal defects), HIV infection, liver disease and certain diet weight loss drugs like fenfluramine and dexfenfluramine.
There are also other causes of secondary pulmonary hypertension, that are not known as pulmonary arterial hypertension. Pulmonary hypertension can be divided into 5 different groups, according to the World Health Organisation (WHO) classification. Pulmonary arterial hypertension classified as WHO Group 1 pulmonary hypertension. There are also WHO Group 2, 3, 4, 5 pulmonary hypertension, that are related to different underlying causes and are treated differently. WHO Group 2 pulmonary hypertension is related to left heart disease (e.g. left sided valve disorder and left sided heart pump failure). WHO group 3 pulmonary hypertension is related to e include lung disorders (emphysema, lung fibrosis, sleep apnea). WHO Group 4 pulmonary hypertension is due to, chronic pulmonary thromboembolism (chronic formation of blood clots in the pulmonary artery). WHO Group 5 pulmonary hypertension is related to haemotological and malignant disorders or endocrine conditions. These different groups of pulmonary hypertension must be clarified by a series of comprehensive investigations before determining diagnosis, risk, prognosis and treatment.
